
Organic Chemistry Text Book (CHEM 3401 and 3402)
11.4 Stereoselective Synthesis.html
Stereoselectivity refers to the preferential formation in a chemical reaction of one product stereoisomer (enantiomer or diastereomer) over another, as a result of inherent reaction specificity, or the influence of chiral features in the substrate, reagent, catalyst or environment. The more specific terms enantioselectivity and diastereoselectivity are commonly used in appropriate situations. When this selectivity results in the formation of an excess of one enantiomer over the other from an achiral or racemic substrate it is sometimes called asymmetric induction.
Any reaction which creates a new stereogenic center may proceed in a stereoselective fashion. The reduction of 3-hexyne to trans-3-hexene by sodium in ammonia, or to cis-3-hexene by Lindlar catalytic hydrogenation are examples. Since the substrate, reagents and products are all achiral, this diastereospecificity lies in the nature of the reactions themselves.
Addition to Carbon-Carbon Double Bonds
Hydroboration
Hydroboration of the prochiral alkene 1-phenylcyclopentene, followed by peroxide oxidation, yields trans-2-phenylcyclopentanol as a racemic mixture. 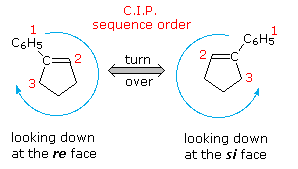 Two new stereogenic centers have been created in this diastereoselective reaction, but since reaction takes place equally at both faces of the double bond, a 50:50 mixture of enantiomers is obtained (equation 1 of the following diagram). This double bond, and others having three different substituents at one carbon, have enantiotopic faces in the same sense that bromochloromethane has a pair of enantiotopic hydrogens. These enantiotopic faces are shown in the diagram on the right. The three different substituents are ranked by the Cahn-Ingold-Prelog sequence rules, and the double bond is viewed from a point above or below its plane. The sequence order is then determined to be a right or left turn (light blue arrow), and the viewing face is given the designation re or si, corresponding to the turn. An addition of a given reagent at the re face will necessarily give the enantiomer of the same addition at the si face, provided the new substituent is not identical to one of the three original groups. Clearly, to make hydroboration an enantioselective reaction, the rate of reaction at one of the faces must be increased over that at the other face. One way of accomplishing this is use an enantiomerically pure chiral alkyl borane reagent for the hydroboration step. The oxidation step is known to proceed with retention of configuration. Several reagents of this kind have been prepared, one useful one being monoisopinocampheylborane (IpcBH2), prepared as shown from the abundant terpene α-pinene. When used for the hydroboration of 1-phenylcyclopentene, the 1R,2S enantiomer is the sole product (equation 2 below). The corresponding diisopinocampheylborane (Ipc2BH) has also been used in similar enantioselective syntheses, but in general the enantiomeric purity of products from reactions with either reagent is seldom as high as the example given here.
Two new stereogenic centers have been created in this diastereoselective reaction, but since reaction takes place equally at both faces of the double bond, a 50:50 mixture of enantiomers is obtained (equation 1 of the following diagram). This double bond, and others having three different substituents at one carbon, have enantiotopic faces in the same sense that bromochloromethane has a pair of enantiotopic hydrogens. These enantiotopic faces are shown in the diagram on the right. The three different substituents are ranked by the Cahn-Ingold-Prelog sequence rules, and the double bond is viewed from a point above or below its plane. The sequence order is then determined to be a right or left turn (light blue arrow), and the viewing face is given the designation re or si, corresponding to the turn. An addition of a given reagent at the re face will necessarily give the enantiomer of the same addition at the si face, provided the new substituent is not identical to one of the three original groups. Clearly, to make hydroboration an enantioselective reaction, the rate of reaction at one of the faces must be increased over that at the other face. One way of accomplishing this is use an enantiomerically pure chiral alkyl borane reagent for the hydroboration step. The oxidation step is known to proceed with retention of configuration. Several reagents of this kind have been prepared, one useful one being monoisopinocampheylborane (IpcBH2), prepared as shown from the abundant terpene α-pinene. When used for the hydroboration of 1-phenylcyclopentene, the 1R,2S enantiomer is the sole product (equation 2 below). The corresponding diisopinocampheylborane (Ipc2BH) has also been used in similar enantioselective syntheses, but in general the enantiomeric purity of products from reactions with either reagent is seldom as high as the example given here.
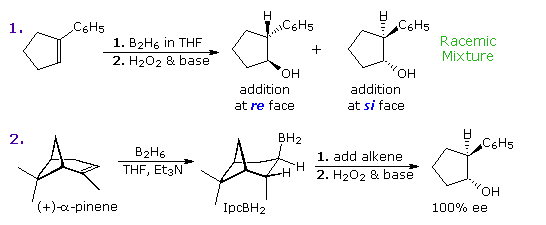
| The ee percentage given in the last equation stands for enantiomeric excess, which is the mole fraction difference in composition of the enantiomers in a mixture. A racemic mixture has equal amounts of enantiomers, so the difference is zero. If there is three times as much (+)-enantiomer as (-)-enantiomer the difference is [0.75 - 0.25 = 0.5] or 50% ee. A pure enantiomer has 100% ee. Differences in enantiomer concentration are usually measured by polarimetry, chiral phase chromatography or nmr with chiral shift reagents. |
Epoxidation
Epoxidation of double bonds has proven to be an effective way of introducing oxygen functionality at both carbon atoms. One or two new stereogenic centers are normally created, often with excellent diastereoselectivity. This transformation is commonly carried out by the action of a peracid (RCO3H), such as peracetic acid or perbenzoic acid, in chloroform or methylene chloride solution. The configuration of double bond substituents (E or Z) is generally preserved, as shown in equation 1 below. Electron donating substituents on the double bond facilitate this reaction, and in the case of deactivation by conjugated electron withdrawing groups, reaction with alkylhydroperoxide anions often achieves epoxidation by a conjugate addition-elimination pathway (equation 2). Steric hindrance usually diverts epoxidation to the less hindered face of a double bond; however hydrogen bonding to a neighboring hydroxyl group may provide a more powerful orienting influence, as in equation 3. In this case note also the selective epoxidation of the more substituted double bond.
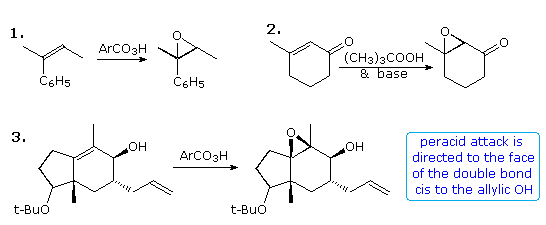
Alternative reaction paths for epoxidation may result in a different diastereoselectivity, as demonstrated above by clicking on the diagram. The trans-fused cycloalkene outlined by the light green box in equation 4 may be epoxidized by a peracid or by base treatment of a bromohydrin intermediate. The upper or β-face of the double bond is blocked by the angular methyl group, so peracid epoxidation occurs at the other face (designated α). Addition of HOBr to the double bond is initiated by electrophilic bromine attack at the less-hindered α-face, and since diaxial addition is favored stereoelectronically, the hydroxyl is bound to the β-face. An Intramolecular SN2 reaction then forms the diastereomeric epoxide. A similar strategy permits diastereoselectivity to be achieved for acyclic alkenes, such as that shown in equation 5. Here a nearby nucleophilic carboxyl group is positioned to reversibly open an iodonium intermediate, producing an iodolactone (central structure). Since this is a reversible reaction, the more stable trans-disubstituted lactone is the major product. Base catalyzed opening of the lactone forms an alkoxide species that immediately displaces iodide to give the epoxide product. Note that all the compounds in this equation are chiral and racemic.
It is known that epoxidation of isolated alkenes by tert-butyl hydroperoxide may be catalyzed by transition metal catalysts, and that an allylic hydroxyl facilitates and facially directs the reaction. By clicking on the above diagram a second time, examples of this reaction will be displayed, with equation 8 illustrating the neighboring group influence. Several features should be noted. First, the Z-configuration of the double bond is preserved in the epoxide products. Second, the allylic hydroxyl group exerts a modest facial diastereoselectivity on the reaction. For clarity the chiral carbinol group is drawn in its R-configuration, but the racemic alcohol would yield the same mixture of diastereomers as racemates.
Professor K. B. Sharpless, Scripps Research Institute, has transformed this general epoxidation reaction into a powerful enantioselective procedure, by the addition of a chiral tartrate ester ligand to a titanium alkoxide catalyst. This important synthetic method is outlined in the following diagram.
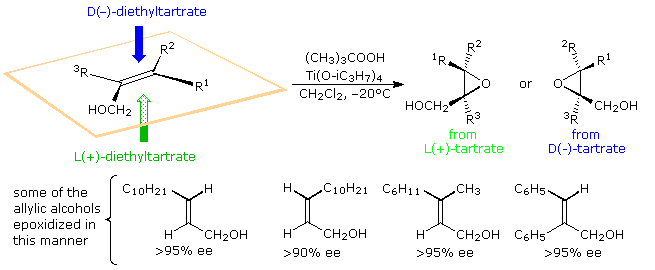
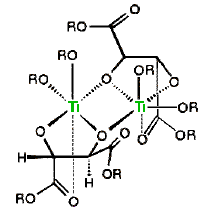 When mixed with one equivalent of diethyl tartrate, titanium tetraisopropoxide forms a dimeric complex with the loss of two isopropyl alcohol molecules. A proposed structure for this complex is shown on the right, with the titanium atoms colored green. Addition of tert-butylhydroperoxide and an allylic alcohol results in the displacement of two more isopropyl alcohols and the formation of a new reactive catalyst complex. A structure for this complex will also be displayed on the right by clicking on the original structure. In this new drawing the double bond of the allylic alcohol is colored blue, and the peroxide oxygens, one of which becomes the epoxide oxygen, are colored red. A pink arrow indicates the bonding of this oxygen to a prochiral face of the double bond. For primary allylic alcohols of the type shown above, Sharpless epoxidation achieves the remarkable conversion of an achiral substrate into a chiral product with high enantioselectivity.
When mixed with one equivalent of diethyl tartrate, titanium tetraisopropoxide forms a dimeric complex with the loss of two isopropyl alcohol molecules. A proposed structure for this complex is shown on the right, with the titanium atoms colored green. Addition of tert-butylhydroperoxide and an allylic alcohol results in the displacement of two more isopropyl alcohols and the formation of a new reactive catalyst complex. A structure for this complex will also be displayed on the right by clicking on the original structure. In this new drawing the double bond of the allylic alcohol is colored blue, and the peroxide oxygens, one of which becomes the epoxide oxygen, are colored red. A pink arrow indicates the bonding of this oxygen to a prochiral face of the double bond. For primary allylic alcohols of the type shown above, Sharpless epoxidation achieves the remarkable conversion of an achiral substrate into a chiral product with high enantioselectivity.
In the reaction of secondary allylic alcohols, where substituent R2 or R3 is an alkyl group (diagram on the right), the allylic alcohol substrate is chiral and the enantiomers react at different rates via diastereomeric transition states. For the racemic alcohol in which R2 (or R3) is a cyclohexyl group, the S-enantiomer reacts over 100 times faster than the R-enantiomer, presumably due to steric hindrance of R2. This rate difference results in a kinetic resolution of this substrate. The S-enantiomer is converted to its erythro diastereomeric epoxide in 98:2 diastereospecificity, while the R-enantiomer is recovered unchanged in over 96% ee.
A model of the Sharpless catalyst may be examined by .
Other catalyst systems for enantioselective epoxidation, as well as hydroxylation and hydrogenation of carbon-carbon double bonds, have been developed and are used in the manufacture of chiral intermediates. The 2001 Nobel Prize in chemistry was awarded to William S. Knowles, Ryoji Noyori, and K. Barry Sharpless for their seminal work in this important field.
Addition to Carbonyl Double Bonds
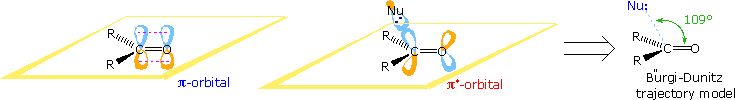
Stereoelectronic factors influence the addition of nucleophilic reagents to carbonyl groups, particularly aldehydes and ketones. In the following diagram the essential molecular orbitals are drawn to the left of the arrow. Initial bonding with a nucleophile is believed to involve the empty antibonding π*-orbital. This explains the favored bonding alignment, known as the Bürgi-Dunitztrajectory.
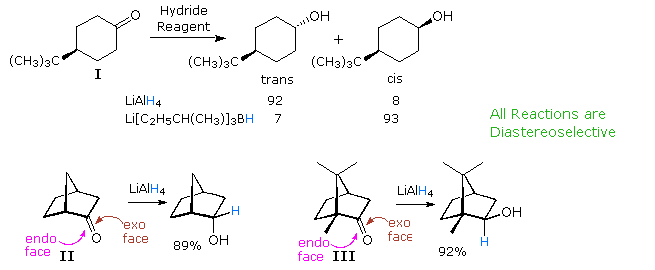
Nucleophilic addition reactions to aldehydes and ketones are probably the earliest and most actively studied examples of stereoselectivity. Selective reduction of 4-tert-butylcyclohexanone (I) to a 10:1 mixture of trans- and cis-4-tert-butylcyclohexanol by LiAlH4 is an example of diastereoselectivity, reflecting a preference for hydride attack at the more hindered axial face of the carbonyl group. This selectivity can be reversed by using a larger hydride reagent, such as Li(sec-butyl)3BH; in which case severe hindrance to axial approach diverts the reaction to the equatorial face. The cis-alcohol is then found to be the major product by over 10:1. Note that in these reactions both the reactants and products are achiral. A similar influence of steric hindrance is seen in reductions of the bicyclo[2.2.1]heptanones II and III (shown below). In compound II the exo face of the prochiral carbonyl group is less hindered than the endo face. The hindrance is reversed in compound III, and the products from LiAlH4 reduction reflect this difference. The relatively fixed configurations of these cyclic ketones permit local stereoelectronic and steric factors to influence reactivity in a well-defined manner.
Models for Addition to Acyclic Substrates
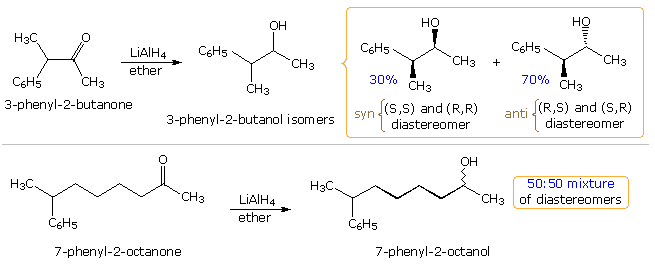
As expected, the behavior of conformationally mobile acyclic compounds is more difficult to rationalize. In the following illustration, two hydride reductions of chiral methyl ketones are shown. Since each ketone has an existing stereogenic site, and since the reduction creates a new chiral center, diastereomeric products are possible. These may be designated in several ways, but the syn-anti notation is generally preferred over the more limited erythro-threo designation. When the existing stereogenic center is located next to the carbonyl group, as in the upper equation, it may influence the proportion of product diastereomers to a significant degree. Because the new stereogenic centers are vicinal, this is termed 1,2-diastereoselectivity, and is the same for both an enantiomerically pure or a racemic reactant. If, however, the stereogenic center is far away from the carbonyl group, it has a negligible influence on the reduction, and a nearly 50:50 mixture of diastereomers is produced (lower example).
A number of models have been proposed to explain the diastereoselectivity of the first reduction. Many conformations about the alpha-C-CO bond may be written, and the challenge is to pick one that accounts for the observed selectivity. Three models will be considered here. For general reference, the substituents on the chiral center adjacent to the carbonyl group are labeled L(large), M (medium) and S (small), reflecting their approximate size. The original structure of these models assumed an orthogonal (90º) approach of the nucleophile to the plane of the carbonyl group, and a reactant-like transition state. In the following diagram this has been changed to the preferred Bürgi-Dunitz approach, as shown by a pink arrow. Each model predicts the correct configuration of the favored diastereomer from LiAlH4 reduction of 3-phenyl-2-butanone (upper equation above with L=C6H5, M=CH3 & S=H). This diastereoselectivity is commonly termed Cram or Felkin selectivity.
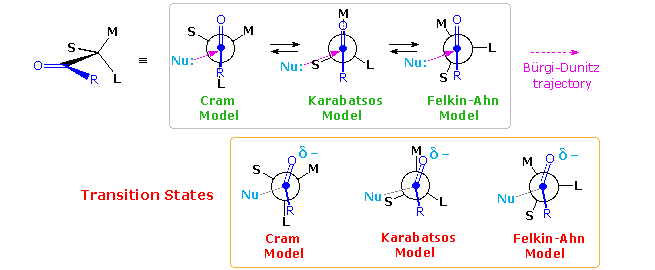
D. J. Cram (Nobel prize 1987) proposed the model on the left in 1952. It suffers from steric hindrance of substituent L with the R substituent on the carbonyl group as well as torsional strain. A more favored conformation was chosen by G. Karabatsos, as shown by the center model. Here the torsional strain is reduced, and the selectivity in reactions of phenylacetaldehydes bearing different α-substituents was rationalized more correctly than by Cram's model. The most recent model (on the right) is that proposed by H. Felkin and elaborated by N.T. Ahn. In this case, overlap of the carbonyl π*-orbital with the C–L σ*-orbital provides electronic stabilization. These models all require classification of S, M & L substituents, occasionally a tricky process, and assume a reactant-like transition state for the reduction. Even so, some bonding of hydride to the carbonyl carbon must take place in the transition state, accompanied by corresponding small structural changes. Plausible structures for these reactant-like transition states are given in the orange box.
A brief analysis of these models is instructive. Although the unfavorable eclipsing of R & L iin the Cram model s slightly relieved in the transition state, the oxygen (and associated metal) moves toward the M group, resulting in increased steric crowding there. The Karabatsos model starts with a favorable conformer, but the nucleophile trajectory nearly eclipses the C-S bond (~20º dihedral angle). The oxygen shift in the transition state relieves eclipsing strain with M, benefiting the transition state energy. Finally, the Felkin-Ahn model seems to offer the best rationalization. The nucleophile trajectory is roughly 40º away from eclipsing the C-S bond, and crowding of both the O⇔M and R⇔S groups is reduced in the transition state.
By clicking on the above diagram a table giving the diastereoselectivity for seven typical addition reactions will appear. The excess of one diastereomer over another is sometimes given as a ratio (e.g. 4:1), but here we use percent diastereomeric excess (%de), defined as the mole fraction difference in diastereomers times 100. Thus, a mixture of 80% diastereomer A and 20% diastereomer B (4:1) has a 60% de. Beneath this table is an example of borohydride reduction of a steroidal methyl ketone (only two rings are shown). The Felkin-Ahn model shown to the right of the equation correctly predicts the diastereoselectivity of the reduction in favor of isomer A. Remarkably, if this ketone is reduced by the bulky borane reagent, disiamylborane (C5H11)2BH, the diastereoselectivity is reversed, with isomer B being formed in 82% de. Since this reduction proceeds by an initial complexing of the Lewis-acidic boron with the carbonyl oxygen, the favored transition state conformer is that in which the small hydrogen and the medium-sized carbon-16 methylene group exchange locations, influenced by the greatly increased size of the oxygen moiety. This change in stereoselectivity is sometimes termed anti-Cram or anti-Felkin selectivity. Aldehydes are especially prone to changes in stereoselectivity for the same reason; however, large nucleophilic reagents favor the Felkin-Ahn product due to reduced hindrance for the Bürgi-Dunitz trajectory (compare cases 1 & 2 in the table).
Because of the conformational mobility of these compounds, it is important to recognize an important precept known as The Curtin-Hammett Principle.
The ratio of products obtained from a a group of equilibrating conformers is determined by transition state energies, not conformer concentrations.
The Chelation Effect
Shortly after proposing his initial model, Cram recognized that ketones and aldehydes having an α-oxygen, nitrogen or sulfur functional group often exhibited a different stereoselectivity. He attributed this behavior to a cyclic chelated conformer, in which the hetero atom of the α-function is coordinated with the metal moiety of the nucleophilic reagent (e.g. Li. Al, B & Mg). The following diagram shows a typical reaction of this kind, in which the chelating ligand is Y and the proposed transition state is enclosed in square brackets. The nucleophile is shown attacking the carbonyl group preferentially from the side of the smallest remaining substituent. Nine examples are listed in the table beneath the equation and two more are shown in the blue tinted box to its right. Powerful coordinating metals such as Zn(II) and Ti(IV) provide the strongest chelation control, as does reaction at a low temperature.
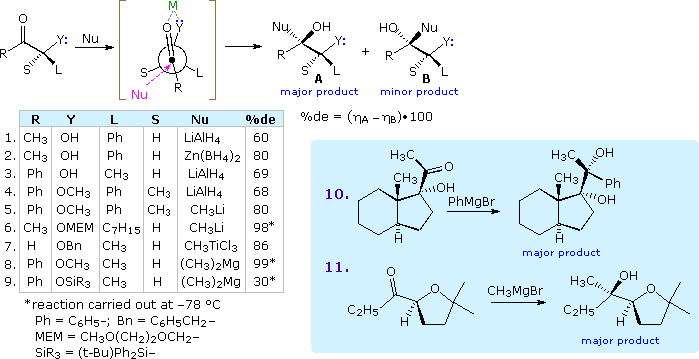
The silyl ether derivative in example 9 is a case of steric hindrance to chelation. The smaller trimethylsilyl ether derivative reacts rapidly and with very high diastereoselectivity, whereas the larger triisopropylsilyl analog reacts slowly and with poor diastereoselectivity favoring isomer B.
Non-chelating Polar Effects
The influence of non-chelating polar substituents on the diastereoselectivity of carbonyl addition reactions proved anomalous. In a study of LiAlH4 reduction of 2-chloropropiophenone J. Cornforth noted that the syn-isomer B was formed in 3:1 ratio with its anti-isomer A, as shown by the top equation in the following diagram. Indications of substituent size from conformational equilibria of substituted cyclohexanes, suggest that a methyl group is significantly larger than a chlorine; consequently, the Felkin model predicts that the anti-isomer should predominate (yellow shaded box). The chelation model leads to a similar prediction. Since chlorine is known to be a poor chelating ligand, Cornforth suggested that dipole repulsion with the carbonyl group (orange arrows) would result in an eclipsed conformation, such as that shown beneath the equation. Such an intermediate predicts the correct diastereoselectivity, but it suffers from the same torsional strain as the Cram model. The Felkin-Ahn model may be modified to reflect the influence of a polar substituent, as shown by the formula right of center. This model also predicts the correct diastereoselectivity, and suggests that additional transition state stabilization may occur by overlap of the approaching nucleophile with the antibonding σ* orbital of the C–Cl bond.
A further test of this rationalization is provided by removing the chelating metal species. Reduction of similar α-substituted propiophenones, C6H5COCHYCH3 (Y = dimethylamino or acetoxy), by the hypervalent hydride reagent, C6H5(CH3)2SiFH(–) (C4H9)4N(+), proceeds with high diastereoselectivity, favoring the syn isomer analogous to B. The absence of a chelating metal combined with the bulk of the hydride donor results in a >95 %de, despite the replacement of chlorine by the much stronger chelating ligands (CH3)2N- and CH3CO2-.
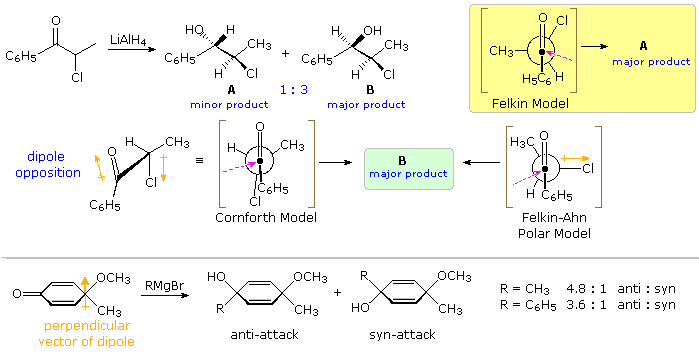
An interesting example in which steric effects and chelation are eliminated is shown at the bottom of the preceding diagram. Here a doubly vinylagous α-substituted ketone undergoes a diastereoselective addition of a Grignard reagent. This and related work by P. Wipf indicate that the selectivity is proportional to the perpendicular component of the molecular dipole, with nucleophile approach favored from the positive side. Curiously, hydride addition occurs without any diastereoselectivity.
1:3-Diastereoselectivity
The previous discussion has focussed on examples of 1:2-diastereoselectivity, where the site of chiral influence has been adjacent to the carbonyl function. If this influence is moved to the β-carbon its strongest effect is transmitted via conformers having six-membered chelate rings. Three examples of such 1:3-diastereoselectivity are shown in the following diagram. For the addition of organometallic nucleophiles a strong coordinating metal like Ti(IV) is needed, both to stabilize the chelate ring and to activate the carbonyl group. As noted in equation 2, the magnesium of a Grignard reagent does not serve this purpose adequately. A possible transition state for the titanium chelated reaction is drawn in the left green shaded box (note the favored axial approach of the nucleophile). Alternatively, intramolecular control may provide selectivity, as shown in equation 3. Sodium triacetoxyborohydride is a weak hydride donor that is commonly used in weakly acidic solutions for reactions such as reductive amination; it does not reduce isolated ketones. Its use in the reduction of 2,6-dimethyl-5-hydroxy-3-heptanone produces excellent diastereoselectivity, as a consequence of the intramolecular hydride transfer intermediate drawn in the right green shaded box (the bulky R substituent prefers to occupy an equatorial-like position).
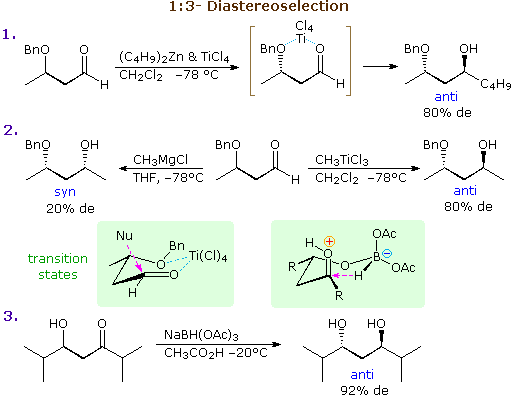
These same factors may also lead to 1:2-diastereoselectivity if α-substitution is present in the substrate. By clicking on the above diagram four examples will be displayed there. In the first two cases the α-carbon is the only stereogenic center, but selectivity occurs because of chelation to a β-functional group. Equations 6 & 7 are additional examples of steric control by intramolecular hydride transfer (see equation 3). Note that the large isopropyl groups control the facial selectivity, so the configuration of the smaller α-methyl substituent is relatively unimportant.
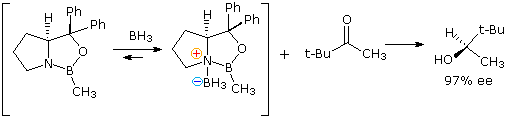
The intramolecular hydride transfer mechanism noted above serves as a model for achieving enantioselective reduction. Several novel catalysts, in which borohydride is complexed with a difunctional chiral ligand, have been developed and used for the enantioselective reduction of prochiral ketones to chiral alcohols. In the following example a chiral ligand prepared from proline is converted to a boron heterocycle. This catalyst binds reversibly with diborane to form the reactive reducing species. Coordination of the ketone oxygen with the Lewis acidic boron orients, and activates the carbonyl group for hydride transfer to its si-face.
Allyl and Crotyl Addition to Aldehydes and Ketones
Allyl Grignard reagents often exhibit different reactivity patterns than similar alkyl reagents, especially with hindered ketones. For example, allylmagnesium bromide adds to diisopropyl ketone as expected, but the major product from reaction with n-propylmagnesium bromide is 2,4-dimethyl-3-pentanol, the result of reduction by a MVP mechanism. Reagents such as methylmagnesium chloride, which have no β-hydrogens to transfer, may act as strong bases and enolize hindered ketones having α-hydrogens, even though the allyl reagent adds in good yield. This characteristic behavior has been attributed to the conjugated (or ambident) nature of this carbon nucleophile, a feature that is readily demonstrated in crotyl (2-butenyl) homologs.
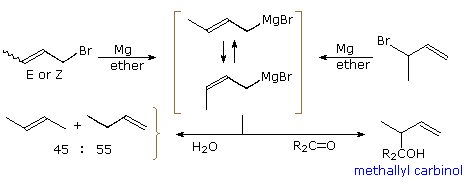
The chemistry of crotyl organometallic reagents has proven both interesting and useful. Conversion of either 1-bromo-2-butene (E or Z), or 3-bromo-1-butene, to a Grignard reagent gives identical mixtures of equilibrating stereo and regioisomers. From nmr evidence, the 1º-isomers drawn within brackets in the following diagram, are the main components (the minor 2º-regioisomers are not drawn). Rapid quenching of this Grignard reagent with water produces a nearly 50:50 mixture of 1- and 2-butene (cis/trans mixture), suggesting facile protonation at both the 1º and 2º allylic sites. Remarkably, simple ketones and aldehydes react exclusively at the 2º-site to form methallyl carbinols. A cyclic transition state for this rearranged addition will be displayed by clicking on the diagram. If the carbonyl group has different substituents, as shown, two new chiral centers are created (red asterisk), and a pair of racemic diastereomers will be formed. Accomplishing this C–C bond formation with high diastereoselectivity would provide chemists with a powerful tool for stereoselective synthesis of complex molecules. Such selectivity would also be different from most of the previous cases, in which diastereoselectivity results from the influence of a preexisting chiral center.
It should be noted that crotyl Grignard additions to very hindered ketones are reversible. The rearranged methallyl carbinol is the kinetically favored product, but heat and extended reaction time leads to formation of the more stable crotyl carbinol, as illustrated by the lower equation in which the magnesium salt of a sterically congested methallyl carbinol is the starting point.
By clicking a second time on the above diagram, some examples of crotyl Grignard addition reactions will be displayed. The greatest product diastereoselectivity is found when one of the carbonyl substituents is much larger than the other. Because both E and Z-isomers of the crotyl reagent are present in the reaction, it is tempting to attribute the formation of the anti-diastereomer to one and the syn-diastereomer to the other. Two such transition states are drawn in the orange box, the E-reagent state leading to the anti-diastereomer and the Z-reagent state to the syn-isomer. At this stage it is not wise to draw such a conclusion, since there are other plausible structures leading to both isomeric products, and according to the Curtin-Hammett principle the favored reaction path among competing possibilities will be that having the lowest activation energy. One additional example will be shown by clicking on the diagram a third time. The steroidal 11-ketone in this example can only react with nucleophiles at its α or ri-face, as a consequence of hindrance by the β-oriented angular methyl groups. Furthermore, the most favored approach of a crotyl moiety appears to be that of the Z-isomer shown in the orange box. Indeed, a single product having the (11R, 20R)-configuration shown was obtained and identified by X-ray analysis. In this case the Z-crotyl reagent produces the anti-diastereomer.
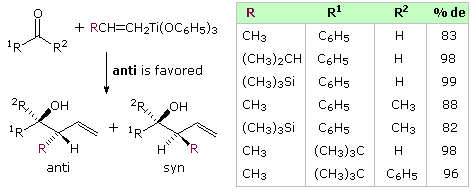 Because of its potential usefulness in synthesis, organic chemists have sought ways to achieve predictable control of diastereoselectivity in the addition of methallyl groups to carbonyl compounds via crotyl reagents. To this end a variety of other metals, ranging from zinc, chromium and titanium to boron, silicon and tin, have been investigated. The zinc, titanium and chromium reagents tend to undergo rapid E/Z interconversion, and either react with mixed diastereoselection or with a strong bias toward anti-selectivity. The examples shown on the right for crotyltitanium triphenoxide illustrate the latter behavior. Similarly, chromium (II) reagents generated in situ from (E or Z)-1-iodo-2-butene add to benzaldehyde with high selectivity favoring the anti-diastereomer.
Because of its potential usefulness in synthesis, organic chemists have sought ways to achieve predictable control of diastereoselectivity in the addition of methallyl groups to carbonyl compounds via crotyl reagents. To this end a variety of other metals, ranging from zinc, chromium and titanium to boron, silicon and tin, have been investigated. The zinc, titanium and chromium reagents tend to undergo rapid E/Z interconversion, and either react with mixed diastereoselection or with a strong bias toward anti-selectivity. The examples shown on the right for crotyltitanium triphenoxide illustrate the latter behavior. Similarly, chromium (II) reagents generated in situ from (E or Z)-1-iodo-2-butene add to benzaldehyde with high selectivity favoring the anti-diastereomer.
Some chair-like cyclic transition states that have been proposed for these reactions will be displayed on the right by clicking on the diagram. The E-based transition state A shown at the top is presumed to be favored, and leads to the anti-product isomer. An alternative Z-transition state, B would form the syn-isomer. The anti-isomer could also be formed by way of the Z-transition state D, drawn in the light gray box. As noted earlier, the assignment of ketone substituents as large, L and small, S, may sometimes be arguable.
Depending on what other ligands are present, crotyl boron, silicon and tin reagents have greater configurational stability than the metal reagents discussed above. Allyl boron and tin reagents may add spontaneously to aldehydes, but allyl silanes generally require catalysis by Lewis acids (this activates the carbonyl function) or by fluoride anions (this increases the nucleophilicity of the allyl moiety).
Addition reactions of a crotylstannane reagent are shown in the following diagram and illustrate some important features. The E and Z isomers of the reagent are relatively stable at room temperature, but react sluggishly with most aldehydes. Chloral, being more electrophilic than most aldehydes, provides a convenient substrate for the uncatalyzed, room temperature addition of this reagent. The stereoselective reactions presented at the top of the diagram suggest a cyclic transition state mechanism in which the E-crotyl reagent gives the anti-diastereomer, and the Z-reagent forms the syn-diastereomer.
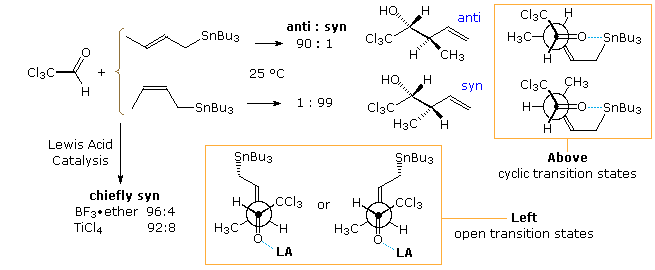
By an initial coordination of the carbonyl oxygen with a Lewis acid, the addition of crotylstannane is accelerated, but both E and Z-isomers form the syn-diastereomer as the major product. Open or acyclic transition states, such as the two drawn at the bottom of the diagram, appear to be functioning in this modification of the reaction. Note that the difference between these E and Z-transition states is very small, and the C–Sn bond is conjugated with the π-orbital of the double bond. Interestingly, if TiCl4 is mixed with the crotyl stannane before it is added to the aldehyde (reverse addition), the anti-isomer of the product is formed preferentially from both E and Z-reagents. A metal exchange reaction, in which the crotylstannane is converted to a crotyltitanium reagent most likely takes place. As noted above, the titanium reagents generally give anti-addition products. Lewis acid catalysis of crotyl silane addition reactions also proceeds by way of an open transition state, and favors the syn-product isomer.
Crotyl Boron Reagents
Although it is possible to prepare E and Z-crotylboranes, CH3CH=CHCH2B(R)2, they interconvert at room temperature. Consequently, any study of addition stereoselectivity for the individual isomers must be conducted at -78 ºC. When this is done the E-isomer favors the anti-product diastereomer, and the Z-isomer favors the syn-product, both with >98% de.
A more convenient approach is to use crotylboronate esters, CH3CH=CHCH2B(OR)2, or crotyltrifluoroborate salts, CH3CH=CHCH2BF3(-) K(+), as the addition reagent. The stereoisomers of these compounds are not only configurationally stable at room temperature, but the compounds themselves are tolerant of exposure to air and moisture, making their preparation storage and use relatively simple. An application using the pinacol ester of crotylboronic acid in shown in the following diagram. The presumed chair-like cyclic transition states for the addition are drawn on the top, with the product from each given underneath. Newman projection drawings are shown in the orange box at the bottom. The data in the table demonstrates the excellent diastereoselection achieved in this reaction.
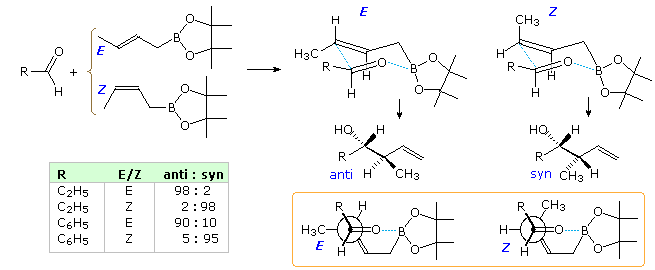
A similar set of equations and data for crotyltrifluoroborate reactions will be displayed above by clicking on the diagram. In this reaction addition is catalyzed by BF3-etherate, which functions as a fluoride ion transfer agent.
Enantioselectivity
When achiral aldehydes and ketones are substrates for addition of allylic reagents, reaction takes place equally at both prochiral faces of the carbonyl double bond. For these addition reactions to be made enantioselective, the rate of reaction at one face of the double bond must be increased over that at the other face. This is the same requirement presented earlier for enantioselective hydroboration, and the isopinocampheyl chiral substituent used by Brown successfully in that case has proven effective here as well. Chiral boronate esters derived from tartrate esters have also served for enantioselective synthesis. Examples of both reagents are given in the following diagram. The specific chiral ligands indicated by the BL2* group are shown in the light green box at the upper right of the diagram. Since both α-pinene and tartaric acid enantiomers (antipodes) are available, the enantioselectivity of these reactions are easily controlled. The top equation shows enantioselective allyl addition to a prochiral aldehyde, creating a single new stereogenic center. The bottom equation shows the analogous crotyl addition in which two new stereogenic centers are formed.
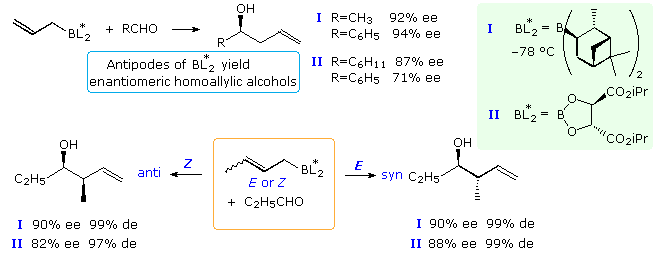
Crotylation of Chiral Aldehydes
The existence of a chiral center alpha to a carbonyl group presents a second, potentially conflicting, factor influencing the stereoselectivity of crotyl addition. The following diagram shows the addition of some achiral allyl boron reagents to three aldehyde substrates of this kind. The first example may be rationalized by the Felkin-Ahn model, the poor stereoselectivity reflecting the similar size of methyl and ethyl groups. The other two examples have polar substituents at the α-carbon, and must be analyzed differently. Because the allyl addition proceeds by a cyclic transition state incorporating a tetra-coordinate boron atom, the chelation model is not an option, and a non-chelation transition state accommodating the polarity of the substituent provides the best explanation. Only modest stereoselectivity takes place.
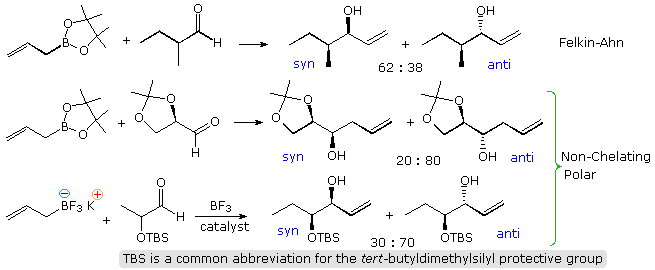
Analogous crotyl additions (both E and Z) have been reported for these aldehyde substrates. The reactions of 2-methylpropanal are shown in the diagram below. The slight preference for the syn-product noted above will be reflected in the 4,5-configuration of the products. The 3,4-configuration is generated by the chair-like cyclic transition states described above, with E-crotyl reagents forming anti diastereomers and Z-reagents giving syn-diastereomers. This selectivity is clearly maintained in all the products, whereas the Felkin-Ahn control of 4,5-configuration is marginal. Fortuitously, the favored transition state for the E-reagent has a Felkin-Ahn configuration, but the Z-transition state does not.
By clicking on the diagram favorable representations of the cyclic transition states for these reactions will be displayed (R=C2H5). Newman projection views of the same transition states are drawn in the orange box.
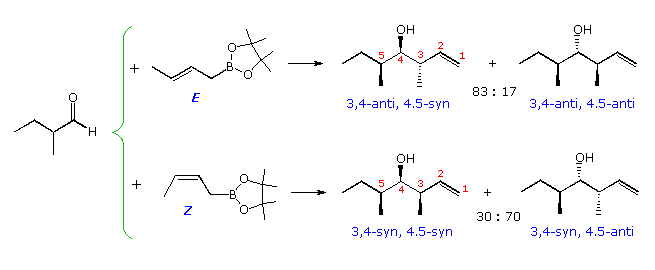
The results of crotyl addition reactions to the two aldehydes having polar α-substituents will be displayed above by clicking a second and third time on the diagram. The dominant stereoselectivity results again from the tendency of E-crotyl reagents to form anti diastereomers and Z-reagents syn-diastereomers. In both cases this is reflected by the 3,4-configuration (note the numbering change for the glyceraldehyde acetal in example 2). The influence of the α-polar substituent is once again less significant, and in these cases reinforcement takes place for the Z-reagent relative to its E-isomer.