Organic Chemistry Text Book (CHEM 3401 and 3402)
21.5.1 Enolate Alkylation
The reaction of alkyl halides with enolate anions presents the same problem of competing SN2 and E2 reaction paths that was encountered earlier in the alkyl halide chapter. Since enolate anions are very strong bases, they will usually cause elimination when reacted with 2º and 3º-halides. Halides that are incapable of elimination and/or have enhanced SN2 reactivity are the best electrophilic reactants for this purpose. Four examples of the C-alkylation of enolate anions in synthesis are displayed in the following diagram. The first two employ the versatile strong base LDA, which is the reagent of choice for most intermolecular alkylations of simple carbonyl compounds. The dichloro alkylating agent used in reaction #1 nicely illustrates the high reactivity of allylic halides and the unreactive nature of vinylic halides in SN2 reactions.
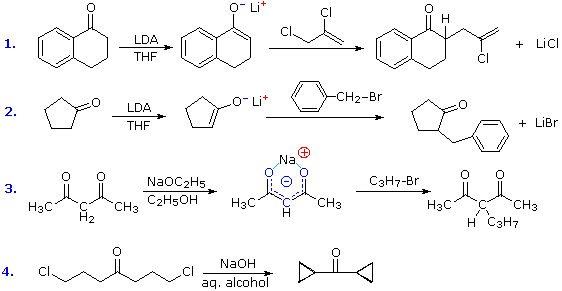
The additive effect of carbonyl groups on alpha-hydrogen acidity is demonstrated by reaction #3. Here the two hydrogen atoms activated by both carbonyl groups are over 1010 times more acidic than the methyl hydrogens on the ends of the carbon chain. Indeed, they are sufficiently acidic (pKa = 9) to allow complete conversion to the enolate anion in aqueous or alcoholic solutions. As shown (in blue), the negative charge of the enolate anion is delocalized over both oxygen atoms and the central carbon. The oxygens are hydrogen bonded to solvent molecules, so the kinetically favored SN2 reaction occurs at the carbon. The monoalkylated product shown in the equation still has an acidic hydrogen on the central carbon, and another alkyl group may be attached there by repeating this sequence.
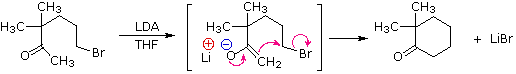
The last example (reaction #4) is an interesting case of intramolecular alkylation of an enolate anion. Since alkylation reactions are irreversible, it is possible to form small highly strained rings if the reactive sites are in close proximity. Reversible bond-forming reactions, such as the aldol reaction, cannot be used for this purpose. The use of aqueous base in this reaction is also remarkable, in view of the very low enolate anion concentration noted earlier for such systems. It is the rapid intramolecular nature of the alkylation that allows these unfavorable conditions to be used.
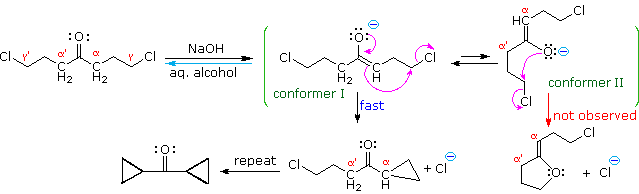
The five-carbon chain of the dichloroketone can adopt many conformations, two of which are approximated in the preceding diagram. Although conformer II of the enolate anion could generate a stable five-membered ring by an intramolecular SN2 reaction, assuming proper orientation of the α and γ' carbon atoms, the concentration of this ideally coiled structure will be very low. In this case O-alkylation of the enolate anion, rather than C-alkylation, is preferred from stereoelectronic arguments (see Baldwin rules). On the other hand, conformations in which the α and γ-carbons are properly aligned for three-membered ring formation are much more numerous, the result being that as fast as the enolate base is formed it undergoes rapid and irreversible cyclization.
Ring closures to four, five, six and seven-membered are also possible by intramolecular enolate alkylation, as illustrated by the following example. In general, five and six-membered rings are thermodynamically most stable, whereas three-membered ring formation is favored kinetically.