
Organic Chemistry Text Book (CHEM 3401 and 3402)
15.1.2b Chemical Shift
Unlike infrared and uv-visible spectroscopy, where absorption peaks are uniquely located by a frequency or wavelength, the location of different nmr resonance signals is dependent on both the external magnetic field strength and the rf frequency. Since no two magnets will have exactly the same field, resonance frequencies will vary accordingly and an alternative method for characterizing and specifying the location of nmr signals is needed. This problem is illustrated by the eleven different compounds shown in the following diagram. Although the eleven resonance signals are distinct and well separated, an unambiguous numerical locator cannot be directly assigned to each.
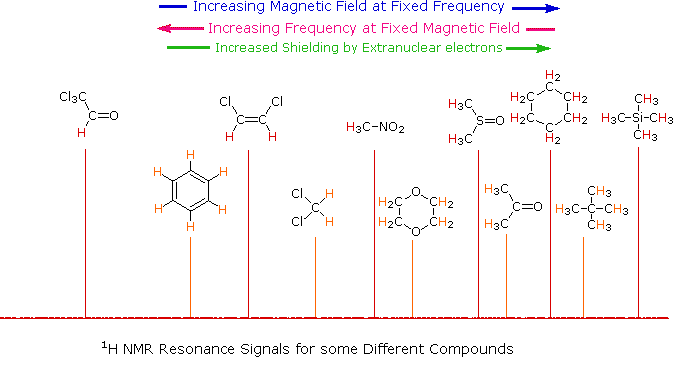
One method of solving this problem is to report the location of an nmr signal in a spectrum relative to a reference signal from a standard compound added to the sample. Such a reference standard should be chemically unreactive, and easily removed from the sample after the measurement. Also, it should give a single sharp nmr signal that does not interfere with the resonances normally observed for organic compounds. Tetramethylsilane, (CH3)4Si, usually referred to as TMS, meets all these characteristics, and has become the reference compound of choice for proton and carbon nmr.
Since the separation (or dispersion) of nmr signals is magnetic field dependent, one additional step must be taken in order to provide an unambiguous location unit. This is illustrated for the acetone, methylene chloride and benzene signals by clicking on the previous diagram. To correct these frequency differences for their field dependence, we divide them by the spectrometer frequency (100 or 500 MHz in the example), as shown in a new display by again clicking on the diagram. The resulting number would be very small, since we are dividing Hz by MHz, so it is multiplied by a million, as shown by the formula in the blue shaded box. Note that νref is the resonant frequency of the reference signal and νsamp is the frequency of the sample signal. This operation gives a locator number called the Chemical Shift, having units of parts-per-million (ppm), and designated by the symbol δ Chemical shifts for all the compounds in the original display will be presented by a third click on the diagram.
The compounds referred to above share two common characteristics:
• The hydrogen atoms in a given molecule are all structurally equivalent, averaged for fast conformational equilibria.
• The compounds are all liquids, save for neopentane which boils at 9 °C and is a liquid in an ice bath.
The first feature assures that each compound gives a single sharp resonance signal. The second allows the pure (neat) substance to be poured into a sample tube and examined in a nmr spectrometer. In order to take the nmr spectra of a solid, it is usually necessary to dissolve it in a suitable solvent. Early studies used carbon tetrachloride for this purpose, since it has no hydrogen that could introduce an interfering signal. Unfortunately, CCl4 is a poor solvent for many polar compounds and is also toxic. Deuterium labeled compounds, such as deuterium oxide (D2O), chloroform-d (DCCl3), benzene-d6 (C6D6), acetone-d6 (CD3COCD3) and DMSO-d6 (CD3SOCD3) are now widely used as nmr solvents. Since the deuterium isotope of hydrogen has a different magnetic moment and spin, it is invisible in a spectrometer tuned to protons.
|
For the properties of some common nmr solvents Click Here. |
|---|
From the previous discussion and examples we may deduce that one factor contributing to chemical shift differences in proton resonance is the inductive effect. If the electron density about a proton nucleus is relatively high, the induced field due to electron motions will be stronger than if the electron density is relatively low. The shielding effect in such high electron density cases will therefore be larger, and a higher external field (Bo) will be needed for the rf energy to excite the nuclear spin. Since silicon is less electronegative than carbon, the electron density about the methyl hydrogens in tetramethylsilane is expected to be greater than the electron density about the methyl hydrogens in neopentane (2,2-dimethylpropane), and the characteristic resonance signal from the silane derivative does indeed lie at a higher magnetic field. Such nuclei are said to be shielded. Elements that are more electronegative than carbon should exert an opposite effect (reduce the electron density); and, as the data in the following tables show, methyl groups bonded to such elements display lower field signals (they are deshielded). The deshielding effect of electron withdrawing groups is roughly proportional to their electronegativity, as shown by the left table. Furthermore, if more than one such group is present, the deshielding is additive (table on the right), and proton resonance is shifted even further downfield.
Proton Chemical Shifts of Methyl Derivatives
|
Proton Chemical Shifts (ppm)
|
|---|
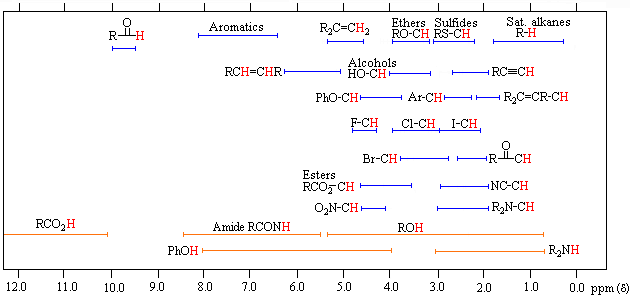
The general distribution of proton chemical shifts associated with different functional groups is summarized in the following chart. Bear in mind that these ranges are approximate, and may not encompass all compounds of a given class. Note also that the ranges specified for OH and NH protons (colored orange) are wider than those for most CH protons. This is due to hydrogen bonding variations at different sample concentrations.
Proton Chemical Shift Ranges* |
||
|---|---|---|
|
Low Field |
|
High Field |
|
* For samples in CDCl3 solution. The δ scale is relative to TMS at δ = 0. |
||
To make use of a calculator that predicts aliphatic proton chemical shifts Click Here. This application was developed at Colby College.