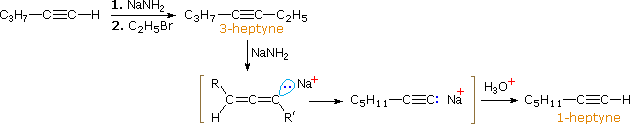Organic Chemistry Text Book (CHEM 3401 and 3402)
9.3 Alkylation of Alkynes
Acidity of Terminal Alkynes
Alkanes are undoubtedly the weakest Brønsted acids commonly encountered in organic chemistry. It is difficult to measure such weak acids, but estimates put the pKa of ethane at about 48. Hybridizing the carbon so as to increase the s-character of the C-H increases the acidity, with the greatest change occurring for the sp-C-H groups found in terminal alkynes. Thus, the pKa of ethene is estimated at 44, and the pKa of ethyne (acetylene) is found to be 25, making it 1023 times stronger an acid than ethane. This increase in acidity permits the isolation of insoluble silver and copper salts of such compounds.
Despite the dramatic increase in acidity of terminal alkynes relative to other hydrocarbons, they are still very weak acids, especially when compared with water, which is roughly a billion times more acidic. If we wish to prepare nucleophilic salts of terminal alkynes for use in synthesis, it will therefore be necessary to use a much stronger base than hydroxide (or ethoxide) anion. Such a base is sodium amide (NaNH2), discussed above, and its reactions with terminal alkynes may be conducted in liquid ammonia or ether as solvents. The products of this acid-base reaction are ammonia and a sodium acetylide salt. Because the acetylide anion is a powerful nucleophile it may displace halide ions from 1º-alkyl halides to give a more highly substituted alkyne as a product (SN2 reaction). This synthesis application is described in the following equations. The first two equations show how acetylene can be converted to propyne; the last two equations present a synthesis of 2-pentyne from propyne.
| H-C≡C-H + NaNH2 (in ammonia or ether) ——> H-C≡C-Na (sodium acetylide) + NH3 |
| H-C≡C-Na + CH3-I ——> H-C≡C-CH3 + NaI |
| CH3-C≡C-H + NaNH2 (in ammonia or ether) ——> CH3-C≡C-Na (sodium propynylide) + NH3 |
| CH3-C≡C-Na + C2H5-Br ——> CH3-C≡C-C2H5 + NaBr |
Because RC≡C:(–) Na(+) is a very strong base (roughly a billion times stronger than NaOH), its use as a nucleophile in SN2 reactions is limited to 1º-alkyl halides; 2º and 3º-alkyl halides undergo elimination by an E2 mechanism.
The enhanced acidity of terminal alkynes relative to alkanes also leads to metal exchange reactions when these compounds are treated with organolithium or Grignard reagents. This exchange, shown below in equation 1, can be interpreted as an acid-base reaction which, as expected, proceeds in the direction of the weaker acid and the weaker base. This factor clearly limits the usefulness of Grignard or lithium reagents when a terminal triple bond is present, as in equation 2.
| 1) RC≡C-H + C2H5MgBr (in ether) ——> RC≡C-MgBr + C2H6 |
| 2) HC≡C-CH2CH2Br + Mg (in ether) ——> [ HC≡C-CH2CH2MgBr] ——> BrMgC≡C-CH2CH2H |
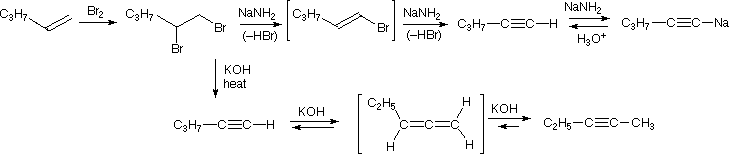
The acidity of terminal alkynes also plays a role in product determination when vicinal (or geminal) dihalides undergo base induced bis-elimination reactions. The following example illustrates eliminations of this kind starting from 1,2-dibromopentane, prepared from 1-pentene by addition of bromine. The initial elimination presumably forms 1-bromo-1-pentene, since base attack at the more acidic and less hindered 1 º-carbon should be favored. The second elimination then produces 1-pentyne. If the very strong base sodium amide is used, the terminal alkyne is trapped as its sodium salt, from which it may be released by mild acid treatment. However, if the weaker base KOH is used for the elimination, the terminal alkyne salt is not formed, or is formed reversibly, and the initially generated 1-pentyne rearranges to the more stable 2-pentyne via an allene intermediate.
In the case of non-terminal alkynes, sodium and potassium amide, and related strong bases from 1 º-amines, are able to abstract protons from carbon atoms adjacent to the triple bond. The resulting allenic carbanions undergo rapid proton transfer equilibria, leading to the relatively stable terminal alkyne conjugate base. This isomerization may be used to prepare longer chain 1-alkynes, as shown in the following conversion of 3-heptyne to 1-heptyne. The R and R' substituents on the allenic intermediate range from propyl to hydrogen, as the proton transfers proceed.