Organic Chemistry Text Book (CHEM 3401 and 3402)
8.5 Halogenation of Alkenes
Addition of Lewis Acids (Electrophilic Reagents)
The proton is not the only electrophilic species that initiates addition reactions to the double bond. Lewis acids like the halogens, boron hydrides and certain transition metal ions are able to bond to the alkene pi-electrons, and the resulting complexes rearrange or are attacked by nucleophiles to give addition products. The electrophilic character of the halogens is well known. Although fluorine is uncontrollably reactive, chlorine, bromine and to a lesser degree iodine react selectively with the double bond of alkenes. The addition of chlorine and bromine to alkenes, as shown in the following general equation, proceeds by an initial electrophilic attack on the pi-electrons of the double bond. Iodine adds reversibly to double bonds, but the equilibrium does not normally favor the addition product, so it is not a useful preparative method. Dihalo-compounds in which the halogens are juxtaposed in the manner shown are called vicinal, from the Latin vicinalis, meaning neighboring.
Other halogen containing reagents which add to double bonds include hypohalous acids, HOX, and sulfenyl chlorides, RSCl. These reagents are unsymmetrical, so their addition to unsymmetrical double bonds may in principle take place in two ways. In practice, these addition reactions are regioselective, with one of the two possible constitutionally isomeric products being favored. The electrophilic moiety of these reagents is the halogen.
(CH3)2C=CH2 + HOBr ——> (CH3)2COH-CH2Br
(CH3)2C=CH2 + C6H5SCl ——> (CH3)2CCl-CH2SC6H5
The regioselectivity of the above reactions may be explained by the same mechanism we used to rationalize the Markovnikov rule. Thus, bonding of an electrophilic species to the double bond of an alkene should result in preferential formation of the more stable (more highly substituted) carbocation, and this intermediate should then combine rapidly with a nucleophilic species to produce the addition product. This is illustrated by the following equation.
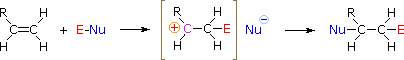
To apply this mechanism we need to determine the electrophilic moiety in each of the reagents. 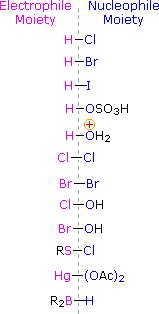 By using electronegativity differences we can dissect common addition reagents into electrophilic and nucleophilic moieties, as shown on the right. In the case of hypochlorous and hypobromous acids (HOX), these weak Brønsted acids (pKa's ca. 8) do not react as proton donors; and since oxygen is more electronegative than chlorine or bromine, the electrophile will be a halide cation. The nucleophilic species that bonds to the intermediate carbocation is then hydroxide ion, or more likely water (the usual solvent for these reagents), and the products are called halohydrins. Sulfenyl chlorides add in the opposite manner because the electrophile is a sulfur cation, RS(+), whereas the nucleophilic moiety is chloride anion (chlorine is more electronegative than sulfur).
By using electronegativity differences we can dissect common addition reagents into electrophilic and nucleophilic moieties, as shown on the right. In the case of hypochlorous and hypobromous acids (HOX), these weak Brønsted acids (pKa's ca. 8) do not react as proton donors; and since oxygen is more electronegative than chlorine or bromine, the electrophile will be a halide cation. The nucleophilic species that bonds to the intermediate carbocation is then hydroxide ion, or more likely water (the usual solvent for these reagents), and the products are called halohydrins. Sulfenyl chlorides add in the opposite manner because the electrophile is a sulfur cation, RS(+), whereas the nucleophilic moiety is chloride anion (chlorine is more electronegative than sulfur).
|
If you understand this mechanism you should be able to write products for the following reactions: |
|
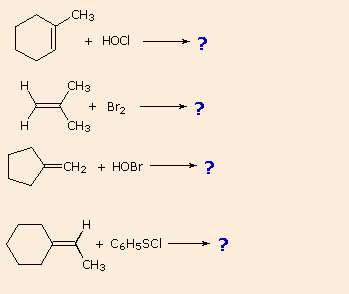 |
|
The addition products formed in reactions of alkenes with mercuric acetate and boron hydrides (compounds shown at the bottom of of the reagent list) are normally not isolated, but instead are converted to alcohols by a substitution reaction. These important synthetic transformations are illustrated for 2-methylpropene by the following equations, in which the electrophilic moiety is colored red and the nucleophile blue. The top reaction sequence illustrates the oxymercuration procedure and the bottom is an example of hydroboration.
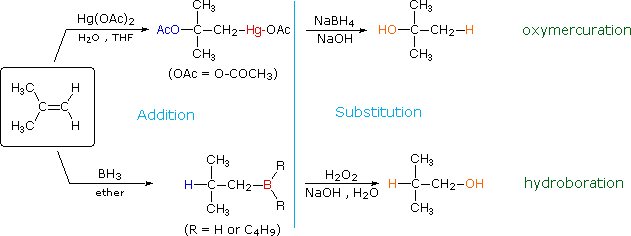
The light blue vertical line separates the addition reaction on the left from the substitution on the right. The atoms or groups that have been added to the original double bond are colored orange in the final product. In both cases the overall reaction is the addition of water to the double bond, but the regioselectivity is reversed. The oxymercuration reaction gives the product predicted by Markovnikov's rule; hydroboration on the other hand gives the "anti-Markovnikov" product. Complementary reactions such as these are important because they allow us to direct a molecular transformation whichever way is desired.
Mercury and boron are removed from the organic substrate in the second step of oxymercuration and hydroboration respectively. These reactions are seldom discussed in detail; however, it is worth noting that the mercury moiety is reduced to metallic mercury by borohydride (probably by way of radical intermediates), and boron is oxidized to borate by the alkaline peroxide. Addition of hydroperoxide anion to the electrophilic borane generates a tetra-coordinate boron peroxide, having the general formula R3B-O-OH(-). This undergoes successive intramolecular shifts of alkyl groups from boron to oxygen, accompanied in each event by additional peroxide addition to electron deficient boron. The retention of configuration of the migrating alkyl group is attributed to the intramolecular nature of the rearrangement.
Since the oxymercuration sequence gives the same hydration product as acid-catalyzed addition of water (see Brønsted acid addition), we might question why this two-step procedure is used at all. The reason lies in the milder reaction conditions used for oxymercuration. The strong acid used for direct hydration may not be tolerated by other functional groups, and in some cases may cause molecular rearrangement (see above).
The addition of borane, BH3, requires additional comment. In pure form this reagent is a dimeric gas B2H6, called diborane, but in ether or THF solution it is dissociated into a solvent coordinated monomer, R2O-BH3. Although diborane itself does not react easily with alkene double bonds, H.C. Brown (Purdue, Nobel Prize 1979) discovered that the solvated monomer adds rapidly under mild conditions. Boron and hydrogen have rather similar electronegativities, with hydrogen being slightly greater, so it is not likely there is significant dipolar character to the B-H bond. Since boron is electron deficient (it does not have a valence shell electron octet) the reagent itself is a Lewis acid and can bond to the pi-electrons of a double bond by displacement of the ether moiety from the solvated monomer. As shown in the following equation, this bonding might generate a dipolar intermediate consisting of a negatively-charged boron and a carbocation. Such a species would not be stable and would rearrange to a neutral product by the shift of a hydride to the carbocation center. Indeed, this hydride shift is believed to occur concurrently with the initial bonding to boron, as shown by the transition state drawn below the equation, so the discrete intermediate shown in the equation is not actually formed. Nevertheless, the carbocation stability rule cited above remains a useful way to predict the products from hydroboration reactions. You may correct the top equation by clicking the button on its right. Note that this addition is unique among those we have discussed, in that it is a single-step process. Also, all three hydrogens in borane are potentially reactive, so that the alkyl borane product from the first addition may serve as the hydroboration reagent for two additional alkene molecules.
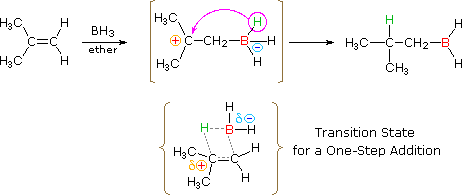 |
To examine models of B2H6. and its dissociation in THF
![]()
Stereoselectivity in Addition Reactions to Double Bonds
As illustrated in the drawing on the right, the pi-bond fixes the carbon-carbon double bond in a planar configuration, and does not permit free rotation about the double bond itself. 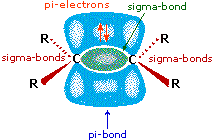 We see then that addition reactions to this function might occur in three different ways, depending on the relative orientation of the atoms or groups that add to the carbons of the double bond: (i) they may bond from the same side, (ii) they may bond from opposite sides, or (iii) they may bond randomly from both sides. The first two possibilities are examples of stereoselectivity, the first being termed syn-addition, and the second anti-addition. Since initial electrophilic attack on the double bond may occur equally well from either side, it is in the second step (or stage) of the reaction (bonding of the nucleophile) that stereoselectivity may be imposed.
We see then that addition reactions to this function might occur in three different ways, depending on the relative orientation of the atoms or groups that add to the carbons of the double bond: (i) they may bond from the same side, (ii) they may bond from opposite sides, or (iii) they may bond randomly from both sides. The first two possibilities are examples of stereoselectivity, the first being termed syn-addition, and the second anti-addition. Since initial electrophilic attack on the double bond may occur equally well from either side, it is in the second step (or stage) of the reaction (bonding of the nucleophile) that stereoselectivity may be imposed.
If the two-step mechanism described above is correct, and if the carbocation intermediate is sufficiently long-lived to freely-rotate about the sigma-bond component of the original double bond, we would expect to find random or non-stereoselective addition in the products. On the other hand, if the intermediate is short-lived and factors such as steric hindrance or neighboring group interactions favor one side in the second step, then stereoselectivity in product formation is likely. The following table summarizes the results obtained from many studies, the formula HX refers to all the strong Brønsted acids. The interesting differences in stereoselectivity noted here provide further insight into the mechanisms of these addition reactions.
| Reagent | H–X | X2 | HO–X | RS–Cl | Hg(OAc)2 | BH3 |
|---|---|---|---|---|---|---|
| Stereoselectivity | mixed | anti | anti | anti | anti | syn |
1. Brønsted Acid Additions
The stereoselectivity of Brønsted acid addition is sensitive to experimental conditions such as temperature and reagent concentration. The selectivity is often anti, but reports of syn selectivity and non-selectivity are not uncommon. Of all the reagents discussed here, these strong acid additions (E = H in the following equation) come closest to proceeding by the proposed two-step mechanism in which a discrete carbocation intermediate is generated in the first step. Such reactions are most prone to rearrangement when this is favored by the alkene structure.
2. Addition Reactions Initiated by Electrophilic Halogen
The halogens chlorine and bromine add rapidly to a wide variety of alkenes without inducing the kinds of structural rearrangements noted for strong acids (first example below). The stereoselectivity of these additions is strongly anti, as shown in many of the following examples.
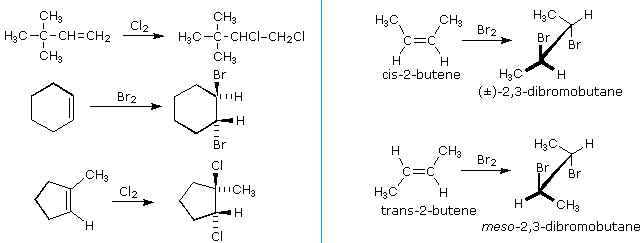
An important principle should be restated at this time. The alkenes shown here are all achiral, but the addition products have chiral centers, and in many cases may exist as enantiomeric stereoisomers. In the absence of chiral catalysts or reagents, reactions of this kind will always give racemic mixtures if the products are enantiomeric. On the other hand, if two chiral centers are formed in the addition the reaction will be diastereomer selective. This is clearly shown by the addition of bromine to the isomeric 2-butenes. Anti-addition to cis-2-butene gives the racemic product, whereas anti-addition to the trans-isomer gives the meso-diastereomer.
We can account both for the high stereoselectivity and the lack of rearrangement in these reactions by proposing a stabilizing interaction between the developing carbocation center and the electron rich halogen atom on the adjacent carbon. This interaction, which is depicted for bromine in the following equation, delocalizes the positive charge on the intermediate and blocks halide ion attack from the syn-location.
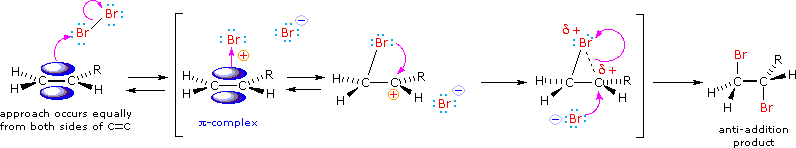
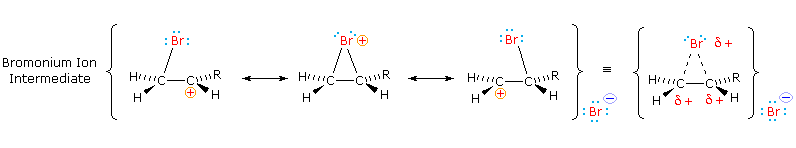
The stabilization provided by this halogen-carbocation bonding makes rearrangement unlikely, and in a few cases three-membered cyclic halonium cations have been isolated and identified as true intermediates. A resonance description of such a bromonium ion intermediate is shown below. The positive charge is delocalized over all the atoms of the ring, but should be concentrated at the more substituted carbon (carbocation stability), and this is the site to which the nucleophile will bond.
|
The stereoselectivity described here is in large part due to a stereoelectronic effect. |
|---|
Because they proceed by way of polar ion-pair intermediates, chlorine and bromine addition reactions are faster in polar solvents than in non-polar solvents, such as hexane or carbon tetrachloride. However, in order to prevent solvent nucleophiles from competing with the halide anion, these non-polar solvents are often selected for these reactions. In water or alcohol solution the nucleophilic solvent may open the bromonium ion intermediate to give an α-halo-alcohol or ether, together with the expected vic-dihalide. Such reactions are sensitive to pH and other factors, so when these products are desired it is necessary to modify the addition reagent. Aqueous chlorine exists as the following equilibrium, Keq ≈ 10-4. By adding AgOH, the concentration of HOCl can be greatly increased, and the chlorohydrin addition product obtained from alkenes.
| Cl2 + H2O |  |
HOCl + HCl |
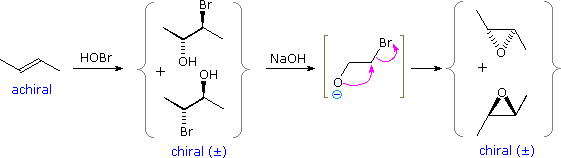
The more widely used HOBr reagent, hypobromous acid, is commonly made by hydrolysis of N-bromoacetamide, as shown below. Both HOCl and HOBr additions occur in an anti fashion, and with the regioselectivity predicted by this mechanism (OH bonds to the more substituted carbon of the alkene).
| CH3CONHBr + H2O |  |
HOBr + CH3CONH2 |
Vicinal halohydrins provide an alternative route for the epoxidation of alkenes over that of reaction with peracids. As illustrated in the following diagram, a base induced intramolecular substitution reaction forms a three-membered cyclic ether called an epoxide. Both the halohydrin formation and halide displacement reactions are stereospecific, so stereoisomerism in the alkene will be reflected in the epoxide product (i.e. trans-2-butene forms a trans-disubstituted epoxide). A general procedure for forming these useful compounds will be discussed in the next section.