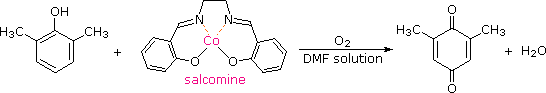Organic Chemistry Text Book (CHEM 3401 and 3402)
12.6.5 Reactions of Phenols
Compounds in which a hydroxyl group is bonded to an aromatic ring are called phenols. The chemical behavior of phenols is different in some respects from that of the alcohols, so it is sensible to treat them as a similar but characteristically distinct group. A corresponding difference in reactivity was observed in comparing aryl halides, such as bromobenzene, with alkyl halides, such as butyl bromide and tert-butyl chloride. Thus, nucleophilic substitution and elimination reactions were common for alkyl halides, but rare with aryl halides. This distinction carries over when comparing alcohols and phenols, so for all practical purposes substitution and/or elimination of the phenolic hydroxyl group does not occur.
1. Acidity of Phenols
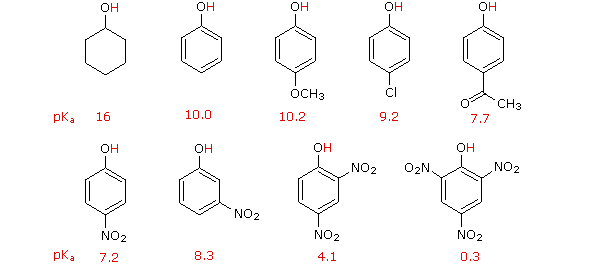
On the other hand, substitution of the hydroxyl hydrogen atom is even more facile with phenols, which are roughly a million times more acidic than equivalent alcohols. This phenolic acidity is further enhanced by electron-withdrawing substituents ortho and para to the hydroxyl group, as displayed in the following diagram. The alcohol cyclohexanol is shown for reference at the top left. It is noteworthy that the influence of a nitro substituent is over ten times stronger in the para-location than it is meta, despite the fact that the latter position is closer to the hydroxyl group. Furthermore additional nitro groups have an additive influence if they are positioned in ortho or para locations. The trinitro compound shown at the lower right is a very strong acid called picric acid.
Why is phenol a much stronger acid than cyclohexanol? To answer this question we must evaluate the manner in which an oxygen substituent interacts with the benzene ring. As noted in our earlier treatment of electrophilic aromatic substitution reactions, an oxygen substituent enhances the reactivity of the ring and favors electrophile attack at ortho and para sites. It was proposed that resonance delocalization of an oxygen non-bonded electron pair into the pi-electron system of the aromatic ring was responsible for this substituent effect. Formulas illustrating this electron delocalization will be displayed when the "Resonance Structures" button beneath the previous diagram is clicked. A similar set of resonance structures for the phenolate anion conjugate base appears below the phenol structures. 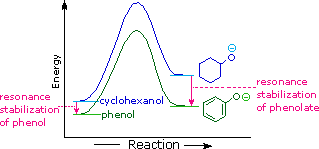
The resonance stabilization in these two cases is very different. An important principle of resonance is that charge separation diminishes the importance of canonical contributors to the resonance hybrid and reduces the overall stabilization. The contributing structures to the phenol hybrid all suffer charge separation, resulting in very modest stabilization of this compound. On the other hand, the phenolate anion is already charged, and the canonical contributors act to disperse the charge, resulting in a substantial stabilization of this species. The conjugate bases of simple alcohols are not stabilized by charge delocalization, so the acidity of these compounds is similar to that of water. An energy diagram showing the effect of resonance on cyclohexanol and phenol acidities is shown on the right. Since the resonance stabilization of the phenolate conjugate base is much greater than the stabilization of phenol itself, the acidity of phenol relative to cyclohexanol is increased. Supporting evidence that the phenolate negative charge is delocalized on the ortho and para carbons of the benzene ring comes from the influence of electron-withdrawing substituents at those sites. The additional resonance stabilization provided by ortho and para nitro substituents will be displayed by clicking the "Resonance Structures" button a second time. You may cycle through these illustrations by repeated clicking of the button.
2. Substitution of the Hydroxyl Hydrogen
As with the alcohols, the phenolic hydroxyl hydrogen is rather easily replaced by other substituents. For example, phenol reacts easily with acetic anhydride to give phenyl acetate. Likewise, the phenolate anion is an effective nucleophile in SN2 reactions, as in the second example below.
 C6H5–O–COCH3 + CH3CO2H
C6H5–O–COCH3 + CH3CO2H
C6H5–O(–) Na(+) + CH3CH2CH3–Br C6H5–O–CH2CH2CH3 + NaBr
3. Electrophilic Substitution of the Phenol Aromatic Ring
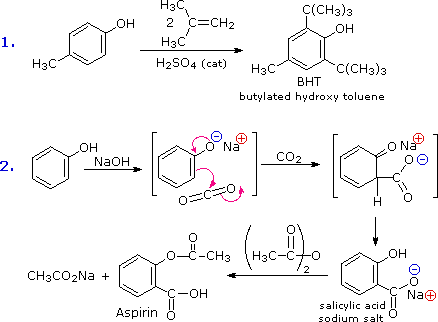
The facility with which the aromatic ring of phenols and phenol ethers undergoes electrophilic substitution has been noted. Two examples are shown in the following diagram. The first shows the Friedel-Crafts synthesis of the food preservative BHT from para-cresol. The second reaction is interesting in that it further demonstrates the delocalization of charge that occurs in the phenolate anion. Carbon dioxide is a weak electrophile and normally does not react with aromatic compounds; however, the negative charge concentration on the phenolate ring enables the carboxylation reaction shown in the second step. The sodium salt of salicylic acid is the major product, and the preference for ortho substitution may reflect the influence of the sodium cation. This is called the Kolbe-Schmidt reaction, and it has served in the preparation of aspirin, as the last step illustrates.
4. Oxidation of Phenols
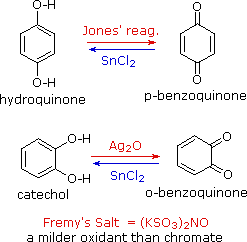
Phenols are rather easily oxidized despite the absence of a hydrogen atom on the hydroxyl bearing carbon. Among the colored products from the oxidation of phenol by chromic acid is the dicarbonyl compound para-benzoquinone (also known as 1,4-benzoquinone or simply quinone); an ortho isomer is also known. These compounds are easily reduced to their dihydroxybenzene analogs, and it is from these compounds that quinones are best prepared. Note that meta-quinones having similar structures do not exist. The redox equilibria between the dihydroxybenzenes hydroquinone and catechol and their quinone oxidation states are so facile that milder oxidants than chromate (Jones reagent) are generally preferred. One such oxidant is Fremy's salt, shown on the right. Reducing agents other than stannous chloride (e.g. NaBH4) may be used for the reverse reaction.
The position of the quinone-hydroquinone redox equilibrium is proportional to the square of the hydrogen ion concentration, as shown by the following half-reactions (electrons are colored blue). The electrode potential for this interconversion may therefore be used to measure the pH of solutions.
Quinone + 2H(+) |
2e(–) –2e(–) |
Hydroquinone |
|---|
Although chromic acid oxidation of phenols having an unsubstituted para-position gives some p-quinone product, the reaction is complex and is not synthetically useful. It has been found that salcomine, a cobalt complex, binds oxygen reversibly in solution, and catalyzes the oxidation of various substituted phenols to the corresponding p-quinones. The structure of salcomine and an example of this reaction are shown in the following equation. The solvent of choice for these oxidations is usually methanol or dimethylformamide (DMF).