
Organic Chemistry Text Book (CHEM 3401 and 3402)
15.1.3 Carbon NMR Spectroscopy
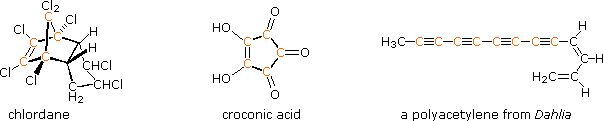
The power and usefulness of 1H nmr spectroscopy as a tool for structural analysis should be evident from the past discussion. Unfortunately, when significant portions of a molecule lack C-H bonds, no information is forthcoming. Examples include polychlorinated compounds such as chlordane, polycarbonyl compounds such as croconic acid, and compounds incorporating triple bonds (structures below, orange colored carbons).
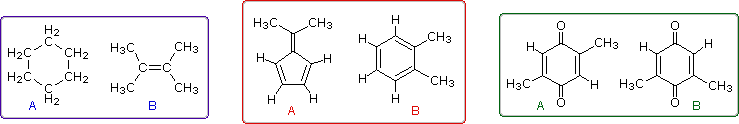
Even when numerous C-H groups are present, an unambiguous interpretation of a proton nmr spectrum may not be possible. The following diagram depicts three pairs of isomers (A & B) which display similar proton nmr spectra. Although a careful determination of chemical shifts should permit the first pair of compounds (blue box) to be distinguished, the second and third cases (red & green boxes) might be difficult to identify by proton nmr alone.
These difficulties would be largely resolved if the carbon atoms of a molecule could be probed by nmr in the same fashion as the hydrogen atoms. Since the major isotope of carbon (12C) has no spin, this option seems unrealistic. Fortunately, 1.1% of elemental carbon is the 13C isotope, which has a spin I = 1/2, so in principle it should be possible to conduct a carbon nmr experiment. It is worth noting here, that if much higher abundances of 13C were naturally present in all carbon compounds, proton nmr would become much more complicated due to large one-bond coupling of 13C and 1H.
|
Many obstacles needed to be overcome before carbon nmr emerged as a routine tool : |
The most important operational technique that has led to successful and routine 13C nmr spectroscopy is the use of high-field pulse technology coupled with broad-band heteronuclear decouplingof all protons. The results of repeated pulse sequences are accumulated to provide improved signal strength. Also, for reasons that go beyond the present treatment, the decoupling irradiation enhances the sensitivity of carbon nuclei bonded to hydrogen.
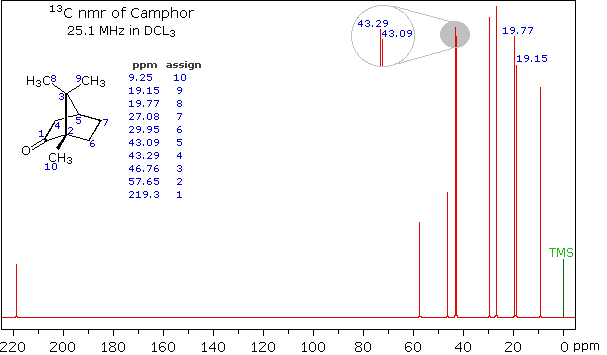
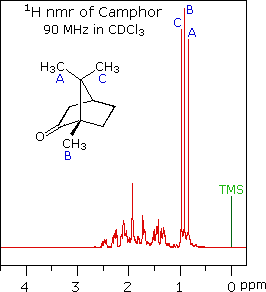
When acquired in this manner, the carbon nmr spectrum of a compound displays a single sharp signal for each structurally distinct carbon atom in a molecule (remember, the proton couplings have been removed). The spectrum of camphor, shown on the left below, is typical. Furthermore, a comparison with the 1H nmr spectrum on the right illustrates some of the advantageous characteristics of carbon nmr. The dispersion of 13C chemical shifts is nearly twenty times greater than that for protons, and this together with the lack of signal splitting makes it more likely that every structurally distinct carbon atom will produce a separate signal. The only clearly identifiable signals in the proton spectrum are those from the methyl groups. The remaining protons have resonance signals between 1.0 and 2.8 ppm from TMS, and they overlap badly thanks to spin-spin splitting.
 |
 |
|---|
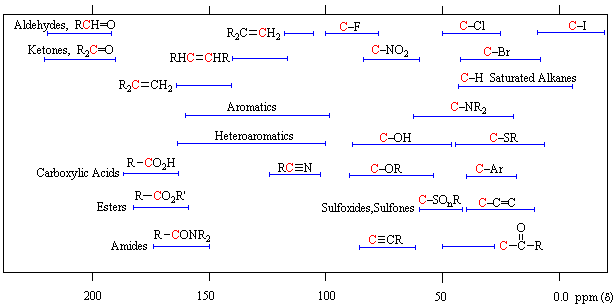
Unlike proton nmr spectroscopy, the relative strength of carbon nmr signals are not normally proportional to the number of atoms generating each one. Because of this, the number of discrete signals and their chemical shifts are the most important pieces of evidence delivered by a carbon spectrum. The general distribution of carbon chemical shifts associated with different functional groups is summarized in the following chart. Bear in mind that these ranges are approximate, and may not encompass all compounds of a given class. Note also that the over 200 ppm range of chemical shifts shown here is much greater than that observed for proton chemical shifts.
13C Chemical Shift Ranges* |
||
|---|---|---|
| Low Field Region |
|
High Field Region |
| * For samples in CDCl3 solution. The δ scale is relative to TMS at δ=0. | ||
The isomeric pairs previously cited as giving very similar proton nmr spectra are now seen to be distinguished by carbon nmr. In the example on the left below (blue box), cyclohexane and 2,3-dimethyl-2-butene both give a single sharp resonance signal in the proton nmr spectrum (the former at δ 1.43 ppm and the latter at 1.64 ppm). However, in its carbon nmr spectrum cyclohexane displays a single signal at δ 27.1 ppm, generated by the equivalent ring carbon atoms (colored blue); whereas the isomeric alkene shows two signals, one at δ 20.4 ppm from the methyl carbons (colored brown), and the other at 123.5 ppm (typical of the green colored sp2 hybrid carbon atoms).
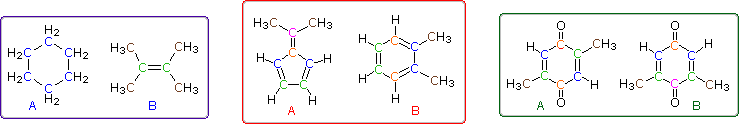
The C8H10 isomers in the center (red) box have pairs of homotopic carbons and hydrogens, so symmetry should simplify their nmr spectra. The fulvene (isomer A) has five structurally different groups of carbon atoms (colored brown, magenta, orange, blue and green respectively) and should display five 13C nmr signals (one near 20 ppm and the other four greater than 100 ppm). Although ortho-xylene (isomer B) will have a proton nmr very similar to isomer A, it should only display four 13C nmr signals, originating from the four different groups of carbon atoms (colored brown, blue, orange and green). The methyl carbon signal will appear at high field (near 20 ppm), and the aromatic ring carbons will all give signals having δ > 100 ppm. Finally, the last isomeric pair, quinones A & B in the green box, are easily distinguished by carbon nmr. Isomer A displays only four carbon nmr signals (δ 15.4, 133.4, 145.8 & 187.9 ppm); whereas, isomer B displays five signals (δ 15.9, 133.3, 145.8, 187.5 & 188.1 ppm), the additional signal coming from the non-identity of the two carbonyl carbon atoms (one colored orange and the other magenta).
|
Variations of the 13C nmr procedure described here can provide additional structural information. |
|---|