
Organic Chemistry Text Book (CHEM 3401 and 3402)
8.6 Dihydroxylation, Epoxidation, and Ozonolysis of Alkenes
Oxidations
(i) Hydroxylation
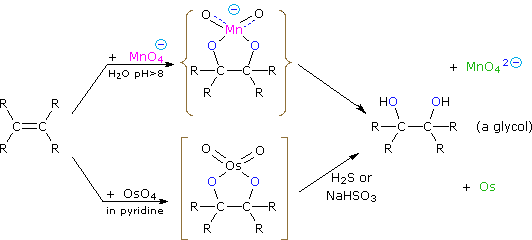
Dihydroxylated products (glycols) are obtained by reaction with aqueous potassium permanganate (pH > 8) or osmium tetroxide in pyridine solution. Both reactions appear to proceed by the same mechanism (shown below); the metallocyclic intermediate may be isolated in the osmium reaction. In basic solution the purple permanganate anion is reduced to the green manganate ion, providing a nice color test for the double bond functional group. From the mechanism shown here we would expect syn-stereoselectivity in the bonding to oxygen, and regioselectivity is not an issue.
When viewed in context with the previously discussed addition reactions, the hydroxylation reaction might seem implausible. Permanganate and osmium tetroxide have similar configurations, in which the metal atom occupies the center of a tetrahedral grouping of negatively charged oxygen atoms. How, then, would such a species interact with the nucleophilic pi-electrons of a double bond? A possible explanation is that an empty d-orbital of the electrophilic metal atom extends well beyond the surrounding oxygen atoms and initiates electron transfer from the double bond to the metal, in much the same fashion noted above for platinum. Back-bonding of the nucleophilic oxygens to the antibonding π*-orbital completes this interaction. The result is formation of a metallocyclic intermediate, as shown below.
(ii) Epoxidation
Some oxidation reactions of alkenes give cyclic ethers in which both carbons of a double bond become bonded to the same oxygen atom. These products are called epoxides or oxiranes. An important method for preparing epoxides is by reaction with peracids, RCO3H. The oxygen-oxygen bond of such peroxide derivatives is not only weak (ca. 35 kcal/mole), but in this case is polarized so that the acyloxy group is negative and the hydroxyl group is positive (recall that the acidity of water is about ten powers of ten weaker than that of a carboxylic acid). If we assume electrophilic character for the OH moiety, the following equation may be written.
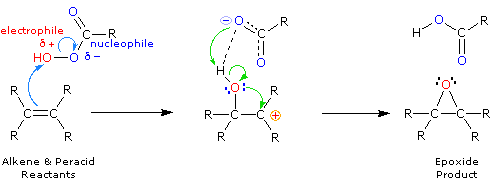 |
It is unlikely that a dipolar intermediate, as shown above, is actually formed. The epoxidation reaction is believed to occur in a single step with a transition state incorporating all of the bonding events shown in the equation. Consequently, epoxidations by peracids always have syn-stereoselectivity, and seldom give structural rearrangement. You may see the transition state by clicking the Change Equation button. Presumably the electron shifts indicated by the blue arrows induce a charge separation that is immediately neutralized by the green arrow electron shifts.
The previous few reactions have been classified as reductions or oxidations, depending on the change in oxidation state of the functional carbons. It is important to remember that whenever an atom or group is reduced, some other atom or group is oxidized, and a balanced equation must balance the electron gain in the reduced species with the electron loss in the oxidized moiety, as well as numbers and kinds of atoms. Starting from an alkene (drawn in the box), the following diagram shows a hydrogenation reaction on the left (the catalyst is not shown) and an epoxidation reaction on the right. Examine these reactions, and for each identify which atoms are reduced and which are oxidized.
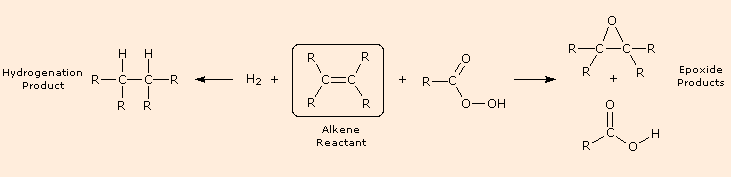 |
Epoxides may be cleaved by aqueous acid to give glycols that are often diastereomeric with those prepared by the syn-hydroxylation reaction described above. Proton transfer from the acid catalyst generates the conjugate acid of the epoxide, which is attacked by nucleophiles such as water in the same way that the cyclic bromonium ion described above undergoes reaction. The result is anti-hydroxylation of the double bond, in contrast to the syn-stereoselectivity of the earlier method. In the following equation this procedure is illustrated for a cis-disubstituted epoxide, which, of course, could be prepared from the corresponding cis-alkene. This hydration of an epoxide does not change the oxidation state of any atoms or groups.
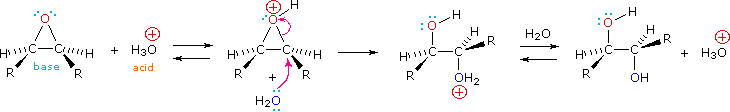
(iii) Oxidative Cleavage of Double Bonds
Ozonolysis
In determining the structural formula of an alkene, it is often necessary to find the location of the double bond within a given carbon framework. One way of accomplishing this would be to selectively break the double bond and mark the carbon atoms that originally formed that bond. For example, there are three isomeric alkenes that all give 2-methylbutane on catalytic hydrogenation. These are 2-methyl-2-butene (compound A), 3-methyl-1-butene (compound B) and 2-methyl-1-butene (compound C), shown in the following diagram. If the double bond is cleaved and the fragments marked at the cleavage sites, the location of the double bond is clearly determined for each case. A reaction that accomplishes this useful transformation is known. It is called ozonolysis, and its application to each of these examples may be seen by clicking the "Show Reaction" button.
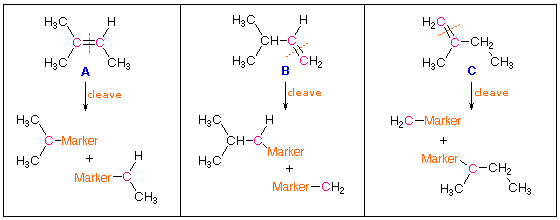 |
Ozone, O3, is an allotrope of oxygen that adds rapidly to carbon-carbon double bonds. Since the overall change in ozonolysis is more complex than a simple addition reaction, its mechanism has been extensively studied. Reactive intermediates called ozonides have been isolated from the interaction of ozone with alkenes, and these unstable compounds may be converted to stable products by either a reductive workup (Zn dust in water or alcohol) or an oxidative workup (hydrogen peroxide). The results of an oxidative workup may be seen by clicking the "Show Reaction" button a second time. Continued clicking of this button repeats the cycle. The chief difference in these conditions is that reductive workup gives an aldehyde product when hydrogen is present on a double bond carbon atom, whereas oxidative workup gives a carboxylic acid or carbon dioxide in such cases. The following equations illustrate ozonide formation, a process that is believed to involve initial syn-addition of ozone, followed by rearrangement of the extremely unstable molozonide addition product. They also show the decomposition of the final ozonide to carbonyl products by either a reductive or oxidative workup.
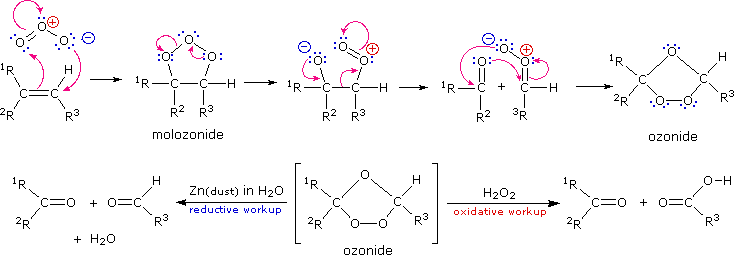
| Ozonolysis To learn more about the mechanism of this fascinating reaction Click Here. |
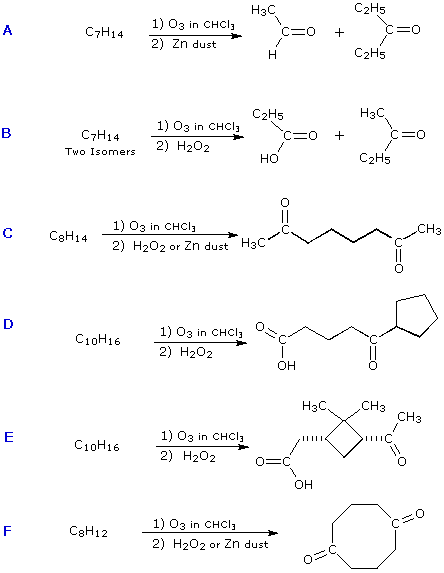
|
||||||
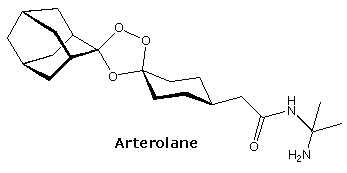
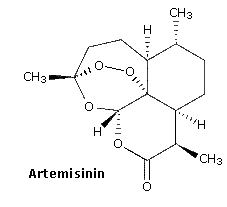
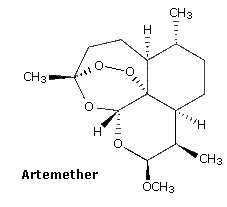
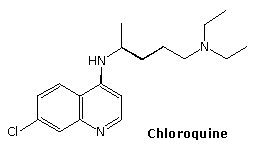
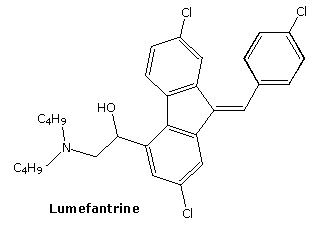
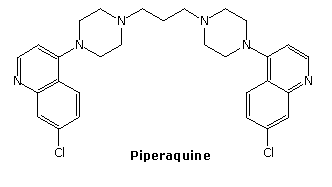
The relatively stable ozonide, arterolane, is a component of a new antimalerial drug, Synriam. Combined with piperaquine, it provides effective treatment of Plasmodium falciparum, the most lethal form of malaria. The ozonide ring is similar to the bridged trioxide unit found in the Chinese herb artemisinin and its derivative artemether, which, combined with lumafantrine have also been used as the antimalerial drug, Coartem. Traditional medications such as chloroquine are effective in only 50 percent of patients where the parasite is drug-resistant.
 |
 |
 |
 |
 |
 |
Glycol Cleavage
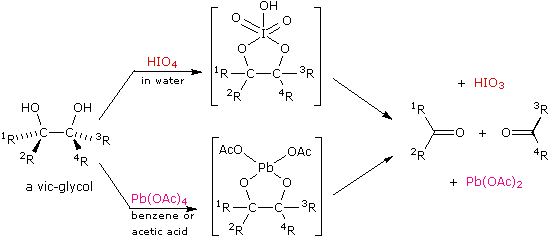
The vicinal glycols prepared by alkene hydroxylation (reaction with osmium tetroxide or permanganate) are cleaved to aldehydes and ketones in high yield by the action of lead tetraacetate (Pb(OAc)4) or periodic acid (HIO4). This oxidative cleavage of a carbon-carbon single bond provides a two-step, high-yield alternative to ozonolysis, that is often preferred for small scale work involving precious compounds. A general equation for these oxidations is shown below. As a rule, cis-glycols react more rapidly than trans-glycols, and there is evidence for the intermediacy of heterocyclic intermediates (as shown), although their formation is not necessary for reaction to occur.
![]()